
Introduzione
Il paracetamolo è uno dei farmaci analgesici e antipiretici più conosciuti e utilizzati al mondo, disponibile senza prescrizione medica, sia in preparazioni mono che multicomponente. In precedenza, si riteneva che il paracetamolo inducesse analgesia inibendo gli enzimi della cicloossigenasi. Recentemente è stato evidenziato che il principale meccanismo analgesico del paracetamolo è la sua metabolizzazione in N-acil fenolammina (AM404), che agisce sui recettori vanilloidi TRPV1 e dei cannabinoidi CB1 nel cervello e nei terminali delle fibre C nel corno dorsale spinale. Pertanto, il paracetamolo induce analgesia agendo non solo sul cervello ma anche sul midollo spinale. Il paracetamolo è un farmaco ben tollerato e produce pochi effetti collaterali gastrointestinali. La deplezione di glutatione, suggerita da alcuni studi, e messa in relazione alla possibile tossicità epatica, non ha evidenze cliniche ai dosaggi raccomandati. Il rischio di effetti collaterali si può manifestare soprattutto se assunto regolarmente a dosi elevate (> 4 g/ die).
Breve storia e meccanismo d’azione
Il paracetamolo è uno dei farmaci più utilizzati al mondo per il trattamento del dolore e della febbre. Come analgesico si distingue dai FANS per l’assenza di attività antinfiammatoria. Rappresenta la terapia analgesica e antipiretica di riferimento anche in età pediatrica, per il favorevole rapporto rischio-beneficio. Occupa una posizione unica tra i farmaci analgesici, sia per la possibilità di impiego in molteplici tipi di dolore che per il profilo di tollerabilità. [1]
Il paracetamolo o acetaminofene (N-acetil-paminofenolo), sintetizzato per la prima volta nel 1877 da Harmon Northrop Morse per riduzione di p-nitrofenolo con stagno in acido acetico glaciale alla Johns Hopkins University, fu introdotto nella pratica clinica come analgesico già nel 1893 (Figura 1).
Figura 1. Harmon Northrop Morse (15 ottobre 1848 – 8 settembre 1920), chimico e docente statunitense, è stato il primo ad aver sintetizzato il paracetamolo. Formula chimica del paracetamolo.
Successivamente venne abbandonato a favore della fenacetina che si rivelò dotata di tossicità soprattutto a livello renale. In realtà, molti anni dopo si scoprì che i benefici effetti prodotti dall’assunzione di fenacetina erano determinati dal fatto che l’organismo trasformava questa molecola in paracetamolo. Era, quindi, il paracetamolo la sostanza che realmente determinava l’analgesia e il calo della temperatura per cui la fenacetina venne abbandonata e il paracetamolo fu reintrodotto nel 1949.
Meccanismo d’azione
Nonostante l’impiego da molti anni, il meccanismo sottostante l’effetto anti-nocicettivo del paracetamolo rimase in gran parte velato nel mistero. Oggi, i meccanismi molecolari attraverso cui paracetamolo agisce sono stati definiti ampiamente. [2] Negli anni sono stati individuati molteplici meccanismi e chiariti i bersagli responsabili dell’attività farmacologica del paracetamolo, che ne corroborano il ruolo antipiretico e analgesico ad azione centrale. È comunemente riconosciuto che il paracetamolo agisce a livello centrale come debole inibitore della sintesi delle prostaglandine (PG) mediante l’azione sulle cicloossigenasi COX-1 e COX-2. Sia in vitro che in vivo è stata osservata una preferenza verso le COX-2, 4 volte superiore alle COX-1. [3]
È interessante notare che il paracetamolo inibisce le COX come i FANS ma non ha attività antinfiammatoria. Le ciclossigenasi, in particolare la prostaglandina H sintasi (PGHS), sono responsabili della sintesi delle prostaglandine e dei trombossani. Le COX sono caratterizzate da due subunità catalitiche, che operano una rispettivamente con funzione ciclossigenasica e l’altra perossidasica. Il paracetamolo, rispetto ai FANS, inibisce la subunità perossidasica, prevenendo la perossidazione delle PGG2 in PGH2. [4,5] All’interno del sito perossidasico delle COX, il paracetamolo agisce come fattore riducente. Nonostante il paracetamolo sia in grado di inibire le ciclossigenasi, a differenza dei FANS, è privo di attività antinfiammatoria. Infatti, nei tessuti periferici e, soprattutto, in corso flogosi, l’elevata concentrazione di idroperossidi endogeni, prodotti dalle cellule infiammatorie, ossidano il radicale Fe nel sito perossidasico, prevenendo l’azione di paracetamolo. Questo meccanismo d’azione spiega perché il paracetamolo non può contrastare l’infiammazione ed avere soltanto un’azione antipiretica e analgesica a livello centrale. [2,4] D’altro canto l’assenza di inibizione delle COX a livello periferico non induce gli effetti collaterali a livello gastrico e renale tipico dei FANS.
Effetto antipiretico
Le prostaglandine svolgono un’azione inibente sui neuroni termosensibili presenti a livello ipotalamico, compromettendo il mantenimento della temperatura corporea a valori fisiologici, con conseguente insorgenza della sintomatologia febbrile. [6] Il paracetamolo agisce a livello del centro termoregolatore ipotalamico, interferendo sulla cascata dell’acido arachidonico e inibendo la produzione delle prostaglandine E2, responsabili dell’innalzamento della temperatura corporea. [7]
Effetto antalgico
Azione indiretta sul sistema degli endocannabinoidi
Studi recenti hanno dimostrato che l’effetto analgesico del paracetamolo è dovuto all’attivazione indiretta dei recettori CB1 dei cannabinoidi. [4] In particolare, il paracetamolo dopo essere stato idrolizzato a p-amminofenolo nel fegato viene coniugato con l’acido arachidonico per azione di un enzima localizzato prevalentemente a livello del sistema nervoso centrale – l’ammide idrolasi degli acidi grassi (FAAH) – a formare il metabolita attivo N-arachidonoilfenolammina (AM404). L’AM404 è un analogo strutturale dell’anandamide e, come tale, è un debole agonista dei recettori cannabinoidi CB1 e CB2 e un inibitore del trasportatore di membrana dell’anandamide. [8] L’AM404, inibendo il reuptake dell’endocannabinoide anandamide dalle strutture presinaptiche, aumenta i livelli di cannabinoidi endogeni e, di conseguenza, l’attivazione dei recettori cannabinoidi sulla membrana post-sinaptica, determinando analgesia.
Attivazione dei recettori vanilloidi TRPV1
Oltre all’inibizione del reuptake degli endocannabinoidi, l’AM404 è in grado attivare i recettori va- nilloidi TRPV1 (Transient Receptor Potential Vanilloid 1). [9] L’AM404 è un potente attivatore dei TRPV1, che a livello della sostanza grigia periacqueduttale determina una stimolazione delle vie inibitorie bulbospinali discendenti, producendo analgesia. [10]
Attivazione dei recettori vanilloidi TRPA1
Un altro metabolita del paracetamolo con proprietà analgesiche è l’N-acetil-p-benzochinoneimina, meglio noto come NAPQI. Il NAPQI è un noto metabolita di paracetamolo, ritenuto mediare gli effetti epatotossici, quando somministrato a dosi superiori a quelle terapeutiche.
A dosi terapeutiche di paracetamolo, il NAPQI si forma anche nel midollo spinale in aree in cui è molto abbondante l’espressione di un altro canale ionico, il TRPA1. Recenti studi hanno dimostrato che gli effetti analgesici del paracetamolo sono anche dovuti all’azione di NAPQI sui canali TRP dei neuroni sensoriali. Infatti, il NAPQI è un potente agonista dei recettori TRPA1. [11]
TRPA1 è un canale cationico non-selettivo espresso nei recettori nocicettivi periferici dei mammiferi e riveste un ruolo fondamentale nella chemonocicezione. È sempre più considerato come un importante obiettivo terapeutico per il trattamento di diverse patologie, tra cui il dolore, l’asma e il prurito cronico. [12] Il TRPA1 è espresso dai neuroni sensoriali primari del corno dorsale del midollo spinale. [13] La sua attivazione provocata dai metaboliti elettrofili del paracetamolo aumenta l’afflusso di calcio, che causa l’inattivazione dei canali del calcio voltaggio-dipendenti (VGCC). L’afflusso di cationi attraverso TRPA1 depolarizza anche la membrana e determina un’inibizione prolungata dei canali del sodio voltaggio-dipendenti (NaV), riducendo così l’eccitabilità neuronale e il rilascio del neurotrasmettitore dipendente dal potenziale d’azione. Ne consegue l’inibizione dell’eccitazione postsinaptica evocata dalla fibra C. [13] L’attivazione dei recettori TRPA1, inoltre, contribuisce all’effetto antipiretico del paracetamolo provocando ipotermia. [11] In conclusione, il paracetamolo attiva direttamente i recettori TRPV1 e TRPA1 e indirettamente i recettori cannabinoidi CB1 aumentando i livelli endogeni di anandamide, entrambi meccanismi presenti nei centri termoregolatori e nei centri del dolore nel cervello. [8]
Oltre a questi meccanismi, altri studi supportano che il paracetamolo agisce anche potenziando le vie inibitorie discendenti serotoninergiche. È stato infatti dimostrato che dopo la somministrazione di paracetamolo si assiste a una significativa down-regulation dei siti di legame per la 5-HT2A a livello della corteccia frontale in riposta al rilascio di 5-HT, dimostrando come il sistema serotoninergico riveste un ruolo nell’effetto analgesico del paracetamolo. [4]
Caratteristiche farmacocinetiche e formulazioni farmaceutiche
Assorbimento
Il paracetamolo somministrato per via orale è ampiamente e rapidamente assorbito. La biodisponibilità orale è intorno al 90%. Il picco di concentrazione plasmatica (Cmax) si realizza tra i 30 e i 60 minuti dall’assunzione. L’assorbimento è prevalente nella prima parte del piccolo intestino e la fase limitante per l’assorbimento è lo svuotamento gastrico nel duodeno. L’assorbimento può variare in modo consistente a seconda della formulazione che viene utilizzata. Compresse e capsule devono disintegrarsi e poi dissolversi, pertanto il tempo per raggiungere il picco di concentrazione è di circa 45-60 minuti dopo la somministrazione. Le compresse effervescenti di paracetamolo e il paracetamolo liquido (elisir, gocce, sciroppo) vengono assorbiti significativamente più velocemente rispetto alle compresse a rilascio regolare. Complessivamente il paracetamolo liquido ha un tempo di picco di circa 30 min. [1,14]
Il paracetamolo mostra un’emivita di 1,25-3 ore, una durata d’azione di 4-6 ore, un basso legame con le proteine plasmatiche (meno del 25%). [15]
Il volume di distribuzione varia da 1 a 2 l/kg negli adulti e da 0,7 a 1 l/kg nei bambini. [1] L’assunzione di paracetamolo a stomaco pieno determina un ritardo nell’assorbimento (Tmax). [16,17] Il cibo riduce, inoltre, la concentrazione massima del paracetamolo somministrato per via orale del 49%. [1] Pertanto, è raccomandabile l’assunzione di paracetamolo a stomaco vuoto. Da notare, tuttavia, che il cibo rallenta l’assorbimento ma non riduce la biodisponibilità totale.
Metabolismo
Il fegato e, in misura minore, il rene e l’intestino sono i principali organi implicati nel metabolismo del paracetamolo. [18] Dopo una dose terapeutica, il paracetamolo viene principalmente trasformato nei coniugati farmacologicamente inattivi glucuronide (paracetamolo glucuronide, 52-57% dei metaboliti urinari) e solfato (paracetamolo solfato, 30-44% dei metaboliti urinari). [18] Solo il 5-10% della dose terapeutica subisce un metabolismo di tipo ossidativo da parte degli enzimi della famiglia del CYP450 2E1 e, in misura minore, 1A2, 3A4 e 2A6, con produzione di un metabolita intermedio altamente reattivo, noto come NAPQI, che può diventare epatotossico in alcune rare condizioni ad alte concentrazioni. [15]
Il NAPQI è normalmente inattivato dalla coniugazione con il glutatione che, successivamente, viene convertito in cisteina o coniugati mercaptici. Questi due metaboliti non risultano tossici e sono facilmente eliminati attraverso le urine e la bile. [8]
Nei bambini la via metabolica predominante è la solfoconiugazione per immaturità del sistema epatico di glucuronazione. I livelli di CYP2E1 sono bassi nei neonati e tendono ad aumentare progressivamente durante il primo anno di età per raggiungere valori prossimi a quelli dell’adulto tra 1-10 anni. I bambini, inoltre, hanno un più rapido turnover del glutatione e, in caso di iperdosaggio, sono meno soggetti alla epatotossicità rispetto agli adulti. [19]
Cambiamenti legati all’età nella farmacocinetica del paracetamolo
Bambini
L’assorbimento per via rettale è considerato irregolare e imprevedibile, con valori riportati di biodisponibilità che vanno dal 24% al 98%. Per la via rettale, il tempo per raggiungere il picco di concentrazione plasmatica varia da 107 a 288 min dopo la somministrazione. [20]
L’età ha anche un impatto sull’assorbimento del paracetamolo. A causa del più lento svuotamento gastrico, l’assorbimento orale nei neonati è ritardato. Le normali condizioni da adulti potrebbero non essere raggiunte prima dei 6-8 mesi di età. [19]
A causa delle differenze nella composizione corporea (es., maggiore contenuto di acqua corporea) e a causa del fatto che il paracetamolo è un composto idrosolubile, il volume di distribuzione è più elevato nel neonato: il volume di distribuzione periferico diminuisce a partire dalle 27 settimane di età post-mestruale (45,0 l × 70 kg - 1) per raggiungere il 110% del suo valore maturo entro i 6 mesi di età. È stato dimostrato che il paracetamolo è in grado di penetrare nel liquido cerebrospinale. È anche in grado di penetrare nella placenta ed è escreto nel latte materno a bassi livelli. [21,22] La dose massima stimata nel neonato attraverso il latte materno è di circa il 2% della dose orale materna di paracetamolo (1,0 g) aggiustata per il peso. [23]
I bambini piccoli sembrano essere più resistenti all’epatotossicità indotta dal paracetamolo a causa sia della riduzione dei tassi di ossidazione da parte del CYP2E1 sia della maggiore capacità del neonato di reintegrare il glutatione rispetto agli adulti. [24]
Gravidanza e allattamento
Sebbene attraversi la barriera placentare, il paracetamolo non ha effetti teratogeni dimostrati a dosi terapeutiche. È, quindi, l’analgesico di scelta in tutte le fasi della gravidanza. La gravidanza può ritardare lo svuotamento gastrico sia per gli effetti meccanici sia per effetti ormonali legati al rilassamento della muscolatura liscia gastrointestinale indotta dal progesterone. Sono state osservate riduzioni dell’area sotto la curva concentrazione-tempo di circa il 20% durante il primo trimestre di gravidanza. Tuttavia, ciò non implica in alcun modo che la dose abituale di paracetamolo debba essere aumentata durante la gravidanza. Le concentrazioni di paracetamolo escrete nel latte materno sono inferiori al 2% della dose ingerita; non vi è, quindi, alcuna controindicazione durante l’allattamento. [23,25,26]
Il paracetamolo è il medicinale più sicuro come analgesico e antipiretico durante la gravidanza. [27,28]
Non esistono farmaci alternativi al paracetamolo; le varie evidenze dei rischi di paracetamolo in gravidanza sono inconcludenti. [29]
Tuttavia, come molti farmaci in gravidanza, il paracetamolo deve essere usato al dosaggio efficace più basso e per il minor tempo possibile. Dovremmo usare il paracetamolo in gravidanza solo quando necessario e non è disponibile alcuna opzione non farmacologica più sicura per alleviare il dolore o la febbre.
Anziani
Diversi studi hanno studiato la farmacocinetica del paracetamolo negli anziani sani, riportando effetti variabili all’età. È stato dimostrato che il paracetamolo viene assorbito rapidamente e completamente dal tratto gastrointestinale, e né la velocità né l’entità dell’assorbimento sembra essere dipendente dall’età. [30]
Il volume di distribuzione diminuisce con l’età e il sesso femminile, il che è coerente con la natura idrofila del farmaco e con i cambiamenti associati all’età nella composizione corporea; non sono state riportate differenze nel volume di distribuzione tra anziani sani e fragili. In generale, l’aumento dell’età non altera la clearance di paracetamolo, che è metabolizzata dal metabolismo coniugativo epatico di fase II. [31]
Ad esempio, uno studio che ha coinvolto 11 donne, di età 89±4 anni, che hanno ricevuto dosi multiple di paracetamolo 1 grammo tre volte al giorno per 5 giorni, non ha mostrato accumulo di farmaco. [32]
Relazione tra farmacocinetica e attività
Alcuni studi riportano che l’effetto analgesico del paracetamolo inizia dopo 30 minuti dall’assunzione. [1,33]
Tuttavia, uno studio randomizzato in doppio cieco ha mostrato un inizio dell’attività analgesica molto più breve. Nello studio è stata confrontata l’efficacia e la sicurezza del paracetamolo 1 g somministrato come iniezione in bolo di 2 minuti o in infusione di 15 minuti con paracetamolo orale 1 g o placebo per l’analgesia in pazienti con dolore da moderato a severo dopo la rimozione del terzo molare incluso. L’inizio dell’analgesia dopo il paracetamolo in vena è stato 3 minuti per la somministrazione in bolo e 5 min per infusione, mentre dopo paracetamolo orale è stata di 11 min (Tabella 1). [34]
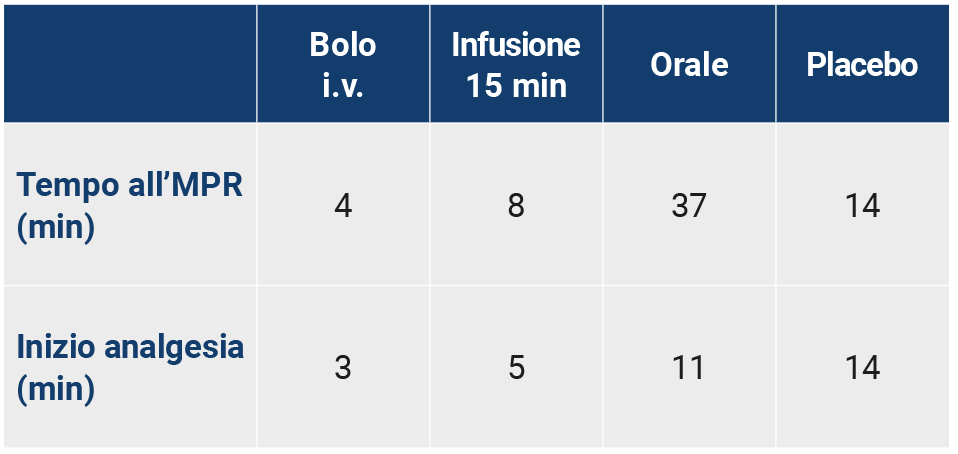
Tabella 1. Inizio della analgesia dopo trattamento con Paracetamolo endovena (2g) o orale (1g) – MPR = significativo sollievo dal dolore (modificata da ref. 34)
Un altro studio, il cui obiettivo principale era quello di verificare la risposta in termini di riduzione del dolore nei pazienti ammessi al pronto soccorso e trattati secondo il protocollo per la gestione infermieristica del dolore nella normale pratica clinica, il 97,4% dei pazienti sono stati trattati con paracetamolo orosolubile 1000 mg. Il tempo medio per la comparsa dell’effetto analgesico è stato di 5,9 minuti (IC 95%: 3,8-8,1). [35]
Interazioni
L’assorbimento per via orale del paracetamolo dipende dalla velocità dello svuotamento gastrico. Pertanto, la somministrazione concomitante di farmaci che rallentano lo svuotamento gastrico (ad es. anticolinergici, oppioidi) riduce l’assorbimento, mentre l’assunzione di procinetici, che aumentano la velocità, può determinarne rispettivamente una diminuzione o un aumento della biodisponibilità.
La somministrazione concomitante di colestiramina riduce l’assorbimento del paracetamolo. L’uso concomitante di paracetamolo con anticoagulanti orali può indurre leggere variazioni nei valori di INR. In questi casi, deve essere condotto un monitoraggio più frequente dei valori di INR durante l’uso concomitante e dopo la sua interruzione. La somministrazione di paracetamolo può interferire con la determinazione della uricemia (mediante il metodo dell’acido fosfotungstico) e con quella della glicemia (mediante il metodo della glucosio-ossidasi-perossidasi). [36]
Tollerabilità e sicurezza
Il paracetamolo è l’antidolorifico da banco e da prescrizione più utilizzato al mondo. [37]
Alla dose terapeutica fino a 3 grammi al giorno per gli adulti il paracetamolo è altamente sicuro. Tuttavia, dosi più alte, accidentali o intenzionali, sono causa di insufficienza epatica acuta. [38]
Il fatto che un farmaco così sicuro possa dare luogo a tossicità da overdose è ampiamente noto.
Il paracetamolo viene metabolizzato per coniugazione con solfato o glucoronidato, che sono inerti e vengono escreti nelle urine. Una frazione modesta di paracetamolo viene convertita in un intermedio tossico altamente reattivo, N-acetil-p-benzochinone immina (NAPQI) da citocromi del complesso P450. Tuttavia, quantità anche sostanziali di NAPQI vengono efficacemente eliminate mediante coniugazione con glutatione (GSH). [39,40]
È evidente, però, che dopo una grande dose di paracetamolo, la reazione di solfonazione si satura e l’accumulo di NAPQI esaurisce il GSH nel fegato, causando un ulteriore accumulo di NAPQI.
Ma quale è la dose di paracetamolo che può rivelarsi altamente tossica?
Studi sugli animali mostrano che occorrono dosi molto alte di paracetamolo per saturare i livelli di GSH. Mitchell et al., hanno misurato il livello di deplezione di GSH nel fegato di topi e la quantità di legame covalente dei metaboliti di paracetamolo radiomarcati (in seguito identificati come NAPQI) nel fegato dopo un’ampia gamma di dosi. Gli autori dello studio hanno mostrato, inoltre, che per causare un esaurimento quasi completo dei livelli di GSH nel fegato occorrono dosi superiori a 400 mg/kg e un forte aumento della quantità di legame covalente. [41,42]
Un modello basato su pazienti dell’Università dello Utah ha mostrato che la dose che determina danno epatico grave e irreversibile è di 20 grammi. Il modello prevede la morte per i pazienti che hanno assunto più di 20 g di paracetamolo senza somministrazione di N-Ac entro le prime 24 ore. [43]
Il dosaggio di 20 grammi è 7 volte la dose giornaliera massima raccomandata che è di 3 grammi.
Considerazione personale è che 7 volte la dose massima raccomandata fa, probabilmente, di ogni farmaco un tossico!
Al dosaggio massimo raccomandato, esiste la possibilità di tossicità epatica da paracetamolo?
Un lavoro relativamente recente ha affrontato il tema della tossicità da paracetamolo in modo analitico. [44] Gli autori mostrano che il loro modello riproduce accuratamente i dati clinici e sperimentali pubblicati sul decorso temporale dose-dipendente del paracetamolo nel plasma, l’accumulo di paracetamolo e dei suoi metaboliti nelle urine e l’esaurimento del glutatione causato dalla coniugazione con il prodotto tossico. Alle dosi terapeutiche, i livelli di NAPQI si mantengono a livelli molto bassi. Inoltre, dimostrano che le dosi terapeutiche riducono il GSH epatico solo di modeste quantità (10%), mentre per arrivare a riduzioni consistenti di GSH bisogna arrivare a sovradosaggi di 10 grammi o più. Inoltre, le dosi terapeutiche croniche riducono il GSH epatico di un valore intorno al 30%. Al termine della terapia, il fegato impiega circa due giorni per sintetizzare una quantità sufficiente di GSH per riportare le concentrazioni alla normalità. Se si considera la capacità antiossidante totale del plasma, dopo paracetamolo somministrato per 14 giorni si assiste ad una riduzione a valori intorno al 10%. Riduzione che è ininfluente sulla capacità antiossidante generale dell’organismo. [45]
Questi studi mostrano chiaramente che alle dosi terapeutiche anche massime non si ha una deplezione di GSH clinicamente significativa. Per avere una deplezione di GSH da parte del paracetamolo che abbia significato clinico occorre andare a dosaggi oltre i 40 mg/kg per somministrazione. L’epatotossicità del paracetamolo è stata esaminata in più studi, sia a dosi terapeutiche sia a sovradosaggio. Ad esempio, in uno studio osservazionale su giovani (18-55 anni), anziani sani (≥ 70 anni) e anziani fragili ricoverati. [42]
I partecipanti hanno ricevuto paracetamolo da 3 a 4 grammi al giorno. Le concentrazioni plasmatiche di alanina aminotransferasi (ALT) misurate al basale e il quinto giorno hanno mostrato variazioni comunque non giudicate patologiche. I pazienti più anziani e, in particolare, i pazienti anziani fragili, hanno avuto una minore incidenza di aumento dell’ALT rispetto ai pazienti più giovani, nonostante avessero concentrazioni medie di paracetamolo significativamente più elevate dopo cinque giorni di trattamento.
Un altro studio ha mostrato che l’incidenza di aumenti significativi dell’alanina aminotransferasi, dopo l’uso quotidiano di paracetamolo come agente analgesico per la chirurgia cardiaca, alla dose di 4 g al giorno, era bassa e principalmente dovuta a complicanze dopo l’intervento chirurgico. Gli autori hanno concluso che i risultati hanno fornito rassicurazioni sulla sicurezza del paracetamolo 4 g al giorno (più alta della dose massima raccomandata) come agente analgesico supplementare per pazienti adulti sottoposti a chirurgia cardiaca. [46]
Gli effetti tossici si osservano quasi sempre a dosi sopra-terapeutiche. Un altro studio prospettico su più di 600 pazienti (età media 37 anni; range 17-76 anni) da 22 centri di assistenza terziaria degli Stati Uniti, il danno epatico indotto da paracetamolo era la principale causa di insufficienza epatica acuta e circa la metà dei casi era dovuta a sovradosaggio non intenzionale. [47]
La dose giornaliera media di paracetamolo nei pazienti che hanno mostrato tossicità era di 7,5 grammi!
Tuttavia, raramente è possibile che alle dosi terapeutiche massime il paracetamolo può causare aumenti transitori degli enzimi epatici e possibilmente epatotossicità, in particolare nelle persone malnutrite (a causa della riduzione del glutatione) e tra coloro che usano induttori degli enzimi epatici (p. es. uso regolare e pesante di alcol, rifampicina, fenitoina, carbamazepina e barbiturici), che aumentano il metabolismo di fase I e le concentrazioni di NAPQI. [48]
Conclusioni
Il paracetamolo è un analgesico efficace, come dimostrato dai numerosi studi disponibili. La ricerca continua sul meccanismo d’azione ha messo in luce che questo farmaco agisce in modo multimodale, sia nel controllo della febbre sia del dolore (Figura 2).
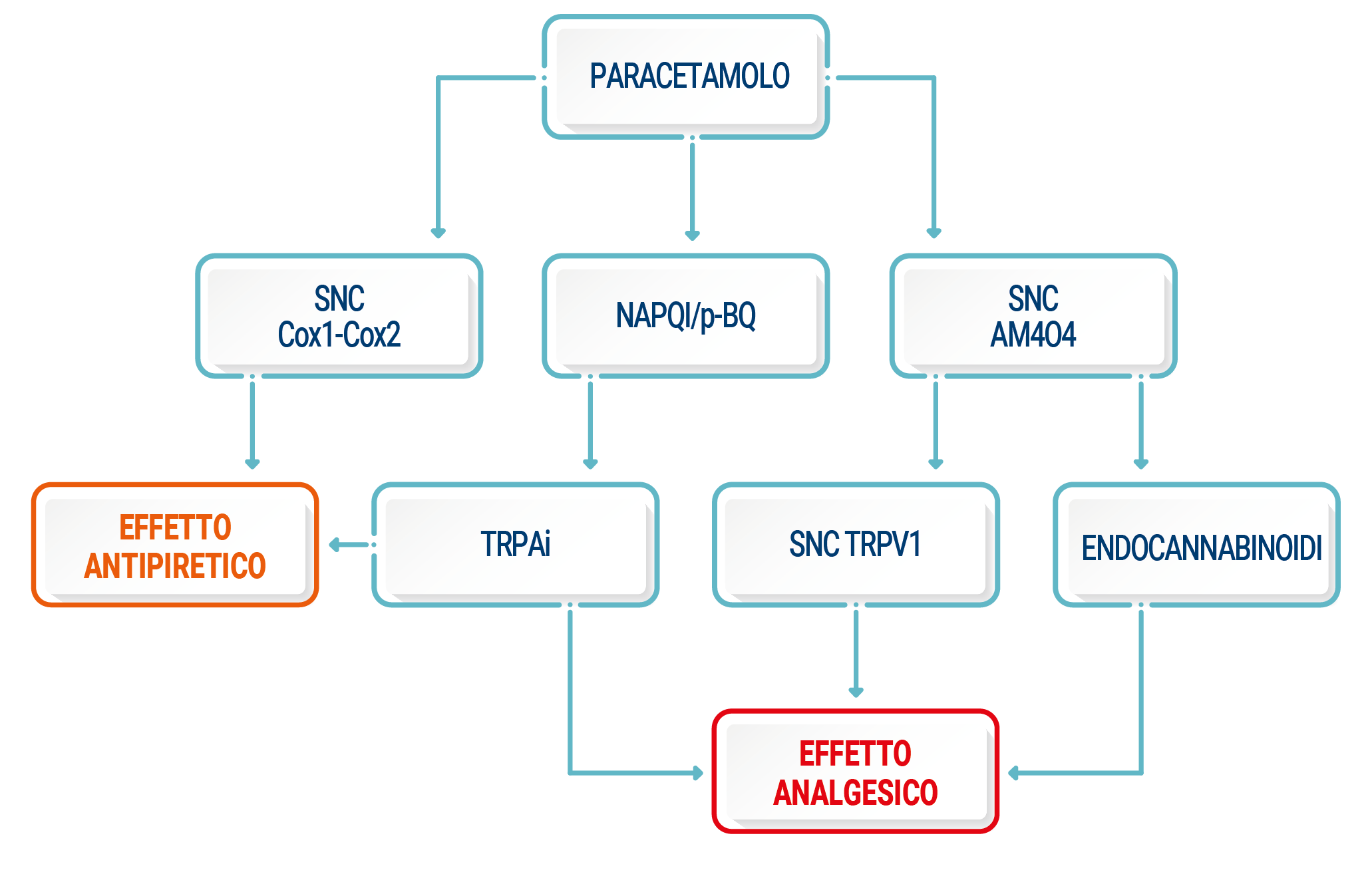
Figura 2. Meccanismo d’azione del paracetamolo. AM404, N-arachidonoilfenolammina; CB1, recettore dei cannabinoidi di tipo 1; SNC, sistema nervoso centrale; NAPQI, cN-acetil-p-benzochinone immina; p-BQ, p-benzochinone; TRPA1, transient receptor potential subfamily ankyrin 1; TRPV1, transient receptor potential subfamily vanilloid-1.
La tollerabilità è eccellente quando usato ai regimi posologici raccomandati. I dubbi emersi in qualche studio sulla possibile deplezione dei livelli di glutatione sono inconsistenti alle posologie raccomandate.
I dati di efficacia, combinati con l’elevata compliance dovuta alle diverse vie di somministrazione disponibili ad un profilo di sicurezza praticamente privo di effetti collaterali clinicamente rilevanti quando vengono seguiti i regimi di dosaggio raccomandati, e il basso costo del paracetamolo suggeriscono che questo farmaco dovrebbe essere usato come analgesico di base di prima linea.
L’Autore ringrazia Angelini Pharma per il contributo incondizionato.
Lascia un commento